






Quantum Chemistry Calculation on Mineral Surface Chemistry Character of Apatite
-
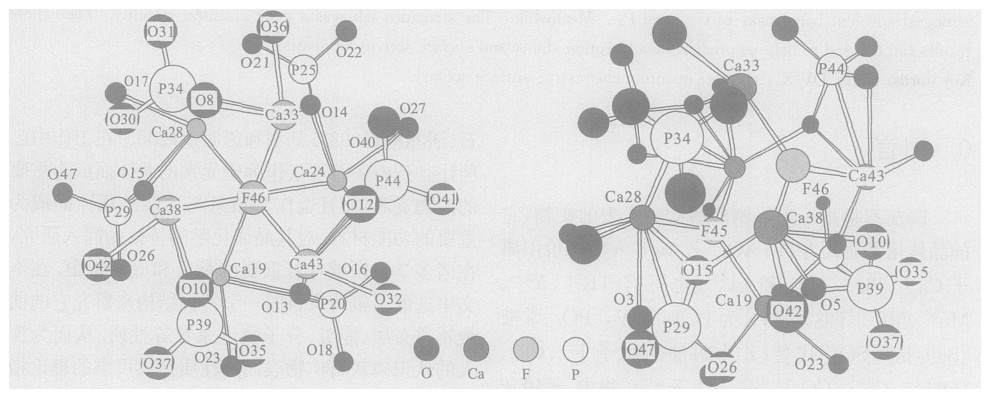
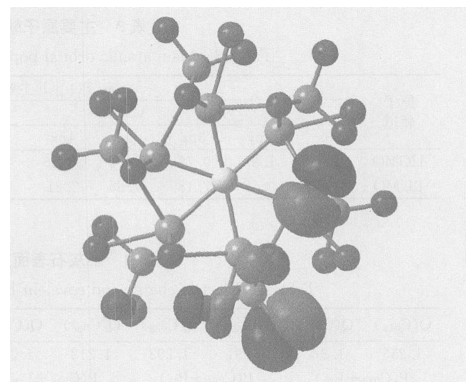
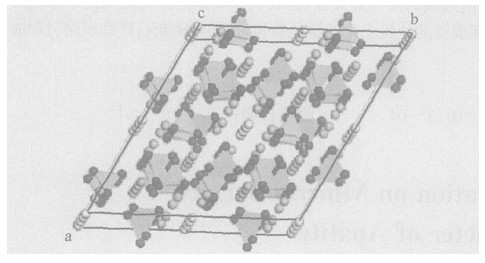
摘要: 采用量子化学从头算起方法中的RHF (HartreeFockRoothaan) 具体方法, 利用STO-3G基组, 对磷灰石矿物表面的能级、前线轨道组成等性质进行计算; 并在磷灰石的特征结构下, 应用DV-Xα法考查了磷灰石的前沿轨道和Fermi能级等性质.通过对计算结果的讨论, 推知在磷灰石的Ca38和P39之间所形成的共价键最强, 也表明该成键的原子之间化学稳定性最强, 同时可以推知在磷灰石表面的其他位置例如Ca24和P25之间成键的位置上化学活性较强, 从而对磷灰石矿物表面吸附能力及表面活性进行研究.Abstract: In this paper, the Gaussian98W software package and the STO-3G basis set have been adopted to study the electric charge, main atomic orbital populations of some frontier molecular orbits and covalent bond level of main atom of apatite surface on the basis of the Hartree-Fock-Roothaan (RHF) method arising from quantum chemistry ab-initio calculation. In apatite crystal structure, the composition characteristics of some frontier molecular orbits and Fermi levels are discussed by means of DV-Xα calculations. The results show that the highest occupied molecular orbits (HOMO) and the lowest unoccupied molecular orbits (LUMO) of the apatite cluster are primarily composed of Ca38 and P39, which results in the strongest covalent bone make of Ca38 and P39. Meanwhile, The structure achieves a good chemical stability. Then these results can be used to help us predict the adsorption ability and surface activity of apatite.
-
Key words:
- DFT /
- DV-Xα /
- apatite /
- quantum chemistry /
- surface activity
-
表 1 磷灰石模型结构中主要的键长和键角
Table 1. Main bond lengths and bond angles of apatite model structure

表 2 磷灰石的前沿分子轨道能量
Table 2. Energies of some frontier molecular orbitals of apatite
(a.u.) 
表 3 主要原子轨道在前沿分子轨道的布居
Table 3. Main atomic orbital populations of some frontier molecular orbitals
(a.u.) 
表 4 磷灰石表面主要原子的电荷和共价键级
Table 4. Electric charge and covalent bond level of main atomic of apatite surface (Au)

-
Andzenlm, J., Wimmer, E., 1992. Density functional Gaussan-type-orbital approach to molecular geometries, vibrtions and reaction energies. J. Chem. Phys. , 96(2): 1280 -1289. doi: 10.1063/1.462165 Hong, H.L., Xiao, R.J., Min, X.M., 2004. Quantum cheistry calculation on the oxidation process of realgar suface. Acta Mineralogica Sinica, 24(1): 95 -98(in Chnese with English abstract). Liu, Y., Peng, M.S., 2003. Advances in the researches on structural substitution of apatite. Acta Petrologica Et Mineralogica, 22(4): 413 -415(in Chinese with Enlish abstract). Pan, Z.L., 1998. crystallography and mineralogy(Third Edtion). Geological Publishing House, Beijing, 224 -226 (in Chinese with English abstract). Xiao, S.X., Wang, C. Y., Chen, T. L., 1988. DFT DV-Xα method applying in chemistry and solid physics. Science Press, Beijing. Yang, H.X., Prewitt, C.T., 1999. On the crystal structure of pseudowollastonite(CaSiO3). American Mineralgist, 84: 929 -932. doi: 10.2138/am-1999-5-628 洪汉烈, 肖睿娟, 闵新民, 2004. 雄黄矿物表面氧化过程的量子化学计算. 矿物学报, 24(1): 95 -98. https://www.cnki.com.cn/Article/CJFDTOTAL-KWXB200401016.htm 刘羽, 彭明生, 2003. 磷灰石结构替换的研究进展. 岩石矿物学杂志, 22(4): 413 -415. https://www.cnki.com.cn/Article/CJFDTOTAL-YSKW200304020.htm 潘兆橹, 1998. 结晶学及矿物学(第三版). 北京: 地质出版社, 224 -226. -
 下载:
下载:
 点击查看大图
点击查看大图
计量
- 文章访问数: 4044
- HTML全文浏览量: 687
- PDF下载量: 13
- 被引次数: 0
